Hipercholesterolemia
Hipercholesterolemia wynika z zaburzeń gospodarki lipidowej prowadzących do podwyższonego stężenia cholesterolu, przede wszystkim frakcji LDL. Może mieć podłoże genetyczne albo rozwijać się pod wpływem czynników środowiskowych i chorób metabolicznych. Hipercholesterolemia przez długi czas pozostaje bezobjawowa, a jej pierwszą kliniczną manifestacją bywają ostre incydenty naczyniowe – choroba wieńcowa, udar mózgu lub niedokrwienie kończyn dolnych. W ciężkich postaciach, zwłaszcza rodzinnych dyslipidemiach, mogą występować zmiany pozanaczyniowe związane z odkładaniem się cholesterolu w tkankach.
e-Konsultacja po Receptę Online
1 Wybierz lek i uzupełnij formularz
2 Przejdź e-konsultację i odbierz zalecenia
3 Możesz otrzymać e-receptę i kod gotowy do realizacji
Leki na hipercholesterolemię
Hipercholesterolemia – przyczyny i objawy. Skąd się bierze i do czego prowadzi wysoki cholesterol?
Najważniejsze informacje
- Hipercholesterolemia to stan, w którym stwierdza się podwyższone stężenie cholesterolu całkowitego i/lub cholesterolu frakcji LDL (tzw. złego cholesterolu) we krwi. Wspomnianym zaburzeniom mogą towarzyszyć także nieprawidłowe wartości innych parametrów lipidowych.
- Przyczyny hipercholesterolemii mogą mieć charakter pierwotny (genetyczny), np. w przypadku rodzinnej hipercholesterolemii, lub wtórny – związany z innymi chorobami, stosowaniem niektórych leków albo czynnikami środowiskowymi i stylem życia.
- Hipercholesterolemia przez wiele lat może przebiegać bezobjawowo, podczas gdy utrzymujące się podwyższone stężenie cholesterolu LDL sprzyja rozwojowi oraz progresji miażdżycy tętnic. Proces ten ma charakter przewlekły i postępujący, a w konsekwencji prowadzi do rozwoju miażdżycowych chorób serca oraz naczyń.
- W wybranych przypadkach, szczególnie w rodzinnej hipercholesterolemii, mogą występować charakterystyczne objawy kliniczne – żółtaki powiek oraz rąbek starczy rogówki.
Hipercholesterolemia wynika z zaburzeń gospodarki lipidowej prowadzących do podwyższonego stężenia cholesterolu, przede wszystkim frakcji LDL. Może mieć podłoże genetyczne albo rozwijać się pod wpływem czynników środowiskowych i chorób metabolicznych. Hipercholesterolemia przez długi czas pozostaje bezobjawowa, a jej pierwszą kliniczną manifestacją bywają ostre incydenty naczyniowe – choroba wieńcowa, udar mózgu lub niedokrwienie kończyn dolnych. W ciężkich postaciach, zwłaszcza rodzinnych dyslipidemiach, mogą występować zmiany pozanaczyniowe związane z odkładaniem się cholesterolu w tkankach.
Cholesterol, LDL, HDL, triglicerydy – za co odpowiadają i jakie funkcje pełnią w organizmie?
Aby lepiej zrozumieć zaburzenia gospodarki lipidowej, warto poznać podstawowe frakcje lipidów obecnych we krwi.
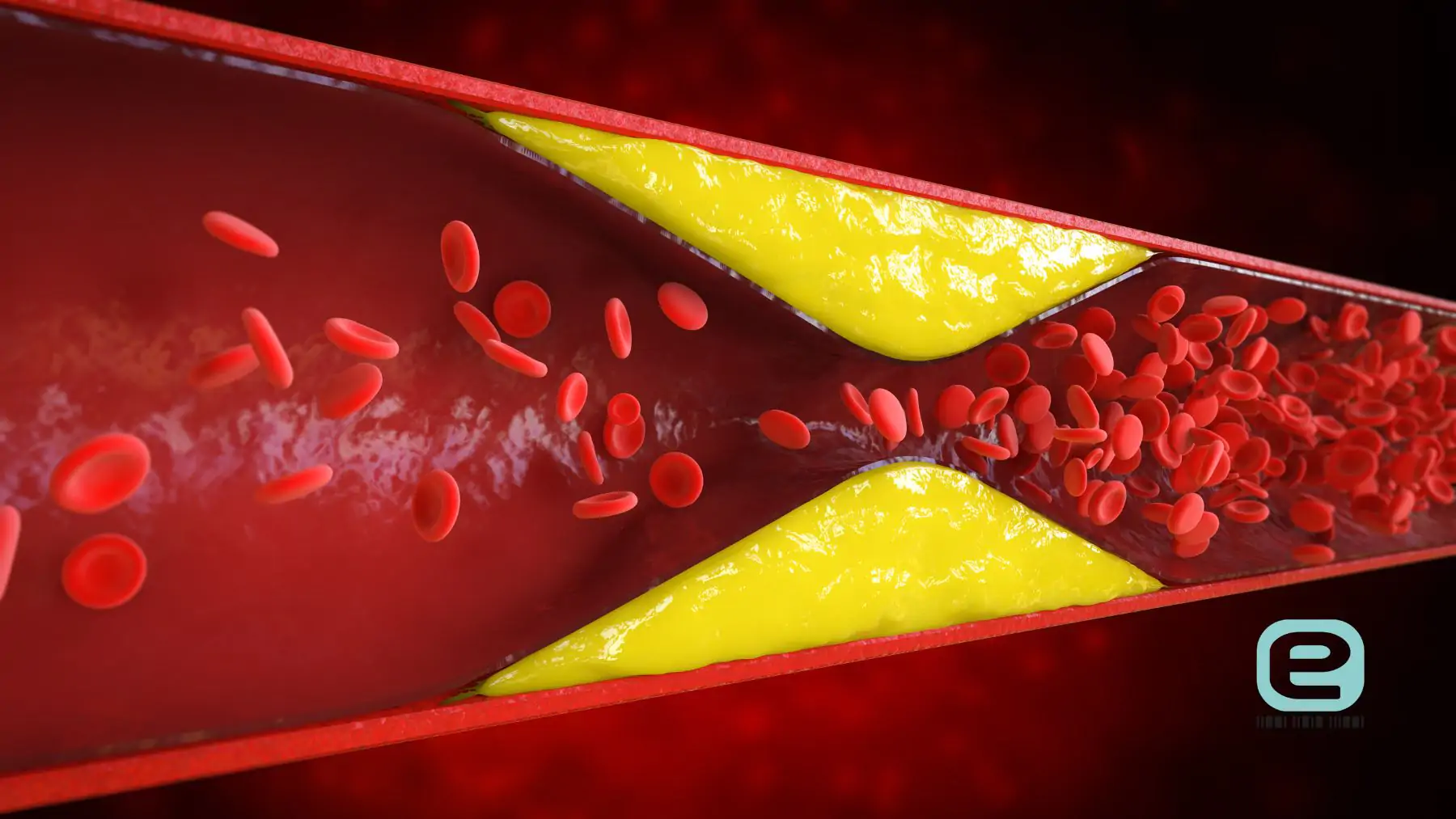
- Cholesterol – jest niezbędny do prawidłowego funkcjonowania organizmu. Bierze udział w budowie błon komórkowych oraz w syntezie hormonów steroidowych, witaminy D i kwasów żółciowych. Większość cholesterolu organizm syntetyzuje samodzielnie, głównie w wątrobie. Problem kliniczny pojawia się wtedy, gdy stężenie cholesterolu LDL jest przewlekle podwyższone, ponieważ sprzyja to rozwojowi miażdżycy. Odkładanie cholesterolu w ścianach tętnic prowadzi do tworzenia blaszek miażdżycowych, które mogą zwężać światło naczyń lub ulegać pęknięciu. Zwiększa to ryzyko zawału serca, udaru mózgu oraz innych chorób sercowo-naczyniowych.
- LDL (lipoproteiny o niskiej gęstości) – odpowiadają za transport cholesterolu z wątroby do tkanek obwodowych. Cholesterol przenoszony przez LDL jest niezbędny m.in. do budowy błon komórkowych oraz syntezy hormonów steroidowych. Problem stanowi jednak przewlekle podwyższone stężenie LDL we krwi. Nadmiar cząsteczek LDL może przenikać do ścian tętnic, gdzie ulega modyfikacjom (m.in. utlenianiu) i uczestniczy w rozwoju stanu zapalnego oraz tworzeniu blaszek miażdżycowych. Proces ten zwiększa ryzyko chorób sercowo-naczyniowych, takich jak zawał serca czy udar mózgu.
- HDL (lipoproteiny o wysokiej gęstości) – uczestniczą w tzw. transporcie zwrotnym cholesterolu – pomagają usuwać jego nadmiar z tkanek oraz ścian naczyń krwionośnych i transportować go do wątroby. Tam cholesterol może zostać wykorzystany, przekształcony lub wydalony z organizmu. Wyższe stężenie HDL zwykle wiąże się z niższym ryzykiem chorób sercowo-naczyniowych, jednak zależność ta nie jest całkowicie jednoznaczna. Sam poziom HDL nie zawsze przekłada się bezpośrednio na działanie ochronne, ponieważ znaczenie ma również funkcjonalność cząsteczek HDL.
- Triglicerydy (TG, trójglicerydy) – stanowią główną formę magazynowania energii w organizmie. Ich stężenie zależy m.in. od diety, spożycia alkoholu, masy ciała, aktywności fizycznej oraz gospodarki węglowodanowej i insulinowej. Podwyższone stężenie triglicerydów często współwystępuje z otyłością, insulinoopornością lub cukrzycą typu 2. Hipertriglicerydemia wiąże się także ze zwiększonym ryzykiem chorób sercowo-naczyniowych, zwłaszcza gdy towarzyszą jej inne zaburzenia metaboliczne. Bardzo wysokie stężenia triglicerydów mogą zwiększać ryzyko ostrego zapalenia trzustki.
Hipercholesterolemia – co to jest?
To zaburzenie gospodarki lipidowej polegające na podwyższonym stężeniu cholesterolu LDL (LDL-C) i/lub cholesterolu całkowitego we krwi w stosunku do wartości referencyjnych dla populacji. Jest to odchylenie biochemiczne, którego znaczenie kliniczne określa się na podstawie całkowitego ryzyka sercowo-naczyniowego oraz obowiązujących celów terapeutycznych.
Przejdź e-konsultację i zapytaj o e-receptę na Twoje leki
Klasyfikacja hipercholesterolemii w ICD-10 i ICD-10-CM
W klasyfikacji ICD-10 stosuje się kod E78.0 (czysta/izolowana hipercholesterolemia), który obejmuje podwyższone stężenie cholesterolu całkowitego i/lub LDL-C bez wyodrębnienia etiologii.
W ICD-10-CM z kolei uwzględnione zostały bardziej szczegółowe kody, m.in. E78.00 (hipercholesterolemia nieokreślona) oraz E78.01 (hipercholesterolemia rodzinna, FH), co pozwala na dokładniejsze różnicowanie postaci klinicznych zaburzenia.
Rodzaje hipercholesterolemii
W zależności od mechanizmu rozwoju wyróżniono:
- hipercholesterolemię pierwotną – uwarunkowaną genetycznie (np. rodzinną hipercholesterolemię), która wynika z mutacji genów wpływających na metabolizm lipoprotein i cholesterolu;
- hipercholesterolemię wtórną – związaną z innymi schorzeniami (np. niedoczynnością tarczycy, zespołem nerczycowym, cholestatycznymi chorobami wątroby, cukrzycą), czynnikami stylu życia (dietą bogatą w tłuszcze nasycone i tłuszcze trans, otyłością, małą aktywnością fizyczną) lub stosowaniem niektórych leków (np. glikokortykosteroidów, retinoidów, cyklosporyny).
Hipercholesterolemia u dzieci i osób młodych
Hipercholesterolemia może występować już w wieku dziecięcym oraz u młodych dorosłych. W takich przypadkach często ma podłoże genetyczne, szczególnie w przebiegu rodzinnej hipercholesterolemii, w której stężenie cholesterolu LDL jest istotnie podwyższone od wczesnych lat życia.
Mimo znacznych zaburzeń lipidowych choroba przez długi czas może pozostawać bezobjawowa, co sprawia, że bywa rozpoznawana dopiero podczas badań laboratoryjnych lub po wystąpieniu przedwczesnych powikłań sercowo-naczyniowych.
Czy hipercholesterolemia jest groźna? Jakie ryzyko niesie dla zdrowia?
Nadmiar cholesterolu LDL sprzyja odkładaniu się lipidów w ścianach tętnic i powstawaniu blaszek miażdżycowych, co prowadzi do stopniowego zwężania naczyń oraz upośledzenia przepływu krwi do narządów. Proces ten zwiększa ryzyko rozwoju miażdżycy oraz jej powikłań sercowo-naczyniowych, takich jak:
- zawał serca;
- udar mózgu;
- choroba tętnic obwodowych;
- niewydolność serca związana z przewlekłym niedokrwieniem.
Ryzyko powikłań wzrasta wraz z czasem utrzymywania się podwyższonego stężenia cholesterolu LDL oraz współistnieniem innych czynników ryzyka sercowo-naczyniowego.
Rozwój miażdżycy w przebiegu hipercholesterolemii – etapy
Hipercholesterolemia jest przewlekłym zaburzeniem metabolicznym rozwijającym się stopniowo – często przez wiele lat, a nawet dekad. Przez długi czas może przebiegać bezobjawowo, mimo że w ścianach naczyń krwionośnych zachodzą już procesy prowadzące do rozwoju miażdżycy. Zjawisko to można opisać etapami.
- Utrzymujące się podwyższone stężenie cholesterolu LDL – nadmiar lipoprotein LDL krąży we krwi i łatwiej przenika do ścian tętnic.
- Dysfunkcja śródbłonka naczyniowego – dochodzi do zaburzenia funkcji wewnętrznej warstwy naczyń krwionośnych, co zwiększa przepuszczalność ściany naczynia i nasila proces zapalny.
- Powstawanie blaszek miażdżycowych – cholesterol odkłada się w ścianach tętnic, prowadząc do tworzenia zmian miażdżycowych i stopniowego pogrubienia ściany naczynia.
- Postępujące zwężenie tętnic – rozwój blaszek miażdżycowych może ograniczać przepływ krwi i upośledzać dopływ tlenu do narządów.
- Wystąpienie objawów klinicznych i powikłań – w zaawansowanym stadium mogą pojawić się symptomy niedokrwienia (choroba wieńcowa, zawał serca, udar mózgu lub choroba tętnic obwodowych).
U części osób pierwszym klinicznym objawem hipercholesterolemii mogą być powikłania sercowo-naczyniowe. Z tego względu regularne wykonywanie lipidogramu odgrywa istotną rolę we wczesnym wykrywaniu zaburzeń gospodarki lipidowej.
Hipercholesterolemia pierwotna (genetyczna)
Wynika z wrodzonych zaburzeń metabolizmu lipidów, najczęściej o podłożu genetycznym. Może być związana z mutacjami genów uczestniczących w wychwycie i usuwaniu cholesterolu LDL z krwi, m.in.: LDLR, APOB oraz PCSK9. Do hipercholesterolemii pierwotnych zalicza się m.in. rodzinną hipercholesterolemię (FH, familial hypercholesterolemia).
Receptomat w telefonie!
Dbaj o zdrowie tak jak Ci wygodnie!Aplikacja Receptomat to innowacyjne rozwiązanie telemedyczne, które pozwala dbać o ciągłość leczenia w prosty sposób. Usługi medyczne są dostępne dla Ciebie przez 7 dni w tygodniu! Umów się na teleporadę, prześlij dokumentację medyczną, ustaw powiadomienia o lekach. Wygodnie, szybko, niezawodnie.
Rodzinna hipercholesterolemia (FH)
Jedna z najczęstszych dziedzicznych chorób wpływających na metabolizm lipidów. Przeważnie dziedziczy się autosomalnie dominująco, co oznacza, że obecność jednej patogennej wersji genu może prowadzić do rozwoju schorzenia.
FH powoduje zwykle znacznie podwyższone stężenie cholesterolu LDL już od dzieciństwa, co istotnie zwiększa ryzyko przedwczesnej miażdżycy oraz chorób sercowo-naczyniowych. Ponadto choroba może przebiegać bezobjawowo przez wiele lat (mimo utrzymujących się zaburzeń lipidowych).
Rozpoznanie opiera się na obrazie klinicznym, wywiadzie rodzinnym, wynikach lipidogramu, a w części przypadków również na badaniach genetycznych.
Postać heterozygotyczna FH
Najczęstsza postać rodzinnej hipercholesterolemii (FH) – występuje, gdy pacjent dziedziczy patogenny wariant genu związanego z rodzinną hipercholesterolemią od jednego z rodziców.
Przebieg choroby jest zwykle łagodniejszy niż w postaci homozygotycznej, jednak nadal wiąże się z istotnie zwiększonym ryzykiem przedwczesnej miażdżycy i chorób sercowo-naczyniowych.
Ze względu na długi okres bezobjawowego lub skąpoobjawowego przebiegu choroba często pozostaje nierozpoznana do czasu wystąpienia powikłań, takich jak: choroba wieńcowa, zawał serca lub udar mózgu.
Postać homozygotyczna FH
Rzadka, ale bardzo ciężka postać rodzinnej hipercholesterolemii. Wiąże się z obecnością patogennych wariantów w obu kopiach genów uczestniczących w metabolizmie cholesterolu LDL. Warianty te mogą zostać odziedziczone od obojga rodziców lub występować w postaci tzw. złożonej heterozygotyczności.
Stężenie cholesterolu LDL osiąga bardzo wysokie wartości już we wczesnym dzieciństwie. Objawy pojawiają się wcześnie i mogą obejmować m.in.: żółtaki ścięgniste, przedwczesną miażdżycę, bóle w klatce piersiowej oraz duszność wysiłkową.
Choroba – jeśli nie zostanie wcześnie rozpoznana i leczona – wiąże się z bardzo wysokim ryzykiem ciężkich powikłań sercowo-naczyniowych w młodym wieku, często już przed 30. rokiem życia.
Hipercholesterolemia wtórna (nabyta)
Jest wynikiem chorób współistniejących, stylu życia lub działania niektórych leków, które prowadzą do wtórnego wzrostu stężenia cholesterolu (najczęściej LDL-C). W przeciwieństwie do hipercholesterolemii pierwotnej nie wynika bezpośrednio z wrodzonych zaburzeń genetycznych metabolizmu lipidów.
W wielu przypadkach stężenie lipidów ulega istotnemu obniżeniu po leczeniu choroby podstawowej, modyfikacji stylu życia lub zmianie stosowanej farmakoterapii, choć efekt ten nie zawsze oznacza pełną normalizację parametrów.
Hipercholesterolemia – przyczyny
Jak wspomniano, hipercholesterolemia może mieć charakter pierwotny (wynikający z uwarunkowań genetycznych zaburzających metabolizm lipoprotein) lub wtórny (związany z chorobami współistniejącymi, stylem życia oraz wpływem niektórych leków).
W praktyce klinicznej oba mechanizmy często współwystępują, co skutkuje kumulacją ryzyka oraz nasileniem zaburzeń lipidowych. Przyczyny wtórne stanowią istotny odsetek przypadków i powinny być rutynowo uwzględniane w diagnostyce różnicowej.
Modyfikowalne i niemodyfikowalne czynniki ryzyka hipercholesterolemii
Ryzyko wystąpienia hipercholesterolemii zależy od czynników niemodyfikowalnych oraz związanych ze stylem życia i środowiskiem metabolicznym.
- Czynniki niemodyfikowalne:
- genetyka – obecność wariantów genetycznych zaburzających metabolizm lipidów, w tym rodzinnej hipercholesterolemii oraz postaci wielogenowych;
- wiek – wraz z wiekiem wzrasta ryzyko zaburzeń lipidowych, co wiąże się ze zmianami metabolicznymi oraz hormonalnymi;
- płeć oraz status hormonalny – u mężczyzn zaburzenia lipidowe i ich następstwa sercowo-naczyniowe pojawiają się statystycznie wcześniej; u kobiet częstość hipercholesterolemii wzrasta po menopauzie, co wiąże się ze spadkiem aktywności estrogenów oraz zmianami w metabolizmie lipoprotein (wzrost LDL, niekiedy spadek HDL).
- Czynniki modyfikowalne:
- dieta bogata w tłuszcze nasycone i trans oraz nadwyżka energetyczna – sprzyjają wzrostowi stężenia LDL;
- niska aktywność fizyczna – wiąże się z pogorszeniem profilu lipidowego, w tym obniżeniem stężenia HDL;
- nadmierna masa ciała – zwiększa ryzyko dyslipidemii;
- palenie tytoniu – obniża stężenie HDL, nasila dysfunkcję śródbłonka i przyspiesza rozwój miażdżycy;
- zaburzenia metaboliczne – w tym zespół metaboliczny i insulinooporność;
- przewlekła ekspozycja na stres – wpływa pośrednio na profil lipidowy (m.in. LDL i HDL) oraz ryzyko miażdżycy.
Przyczyny monogenowe rodzinnej hipercholesterolemii
Postać monogenowa wynika z mutacji pojedynczego genu i zwykle wiąże się z podwyższonym stężeniem LDL-cholesterolu oraz zwiększonym ryzykiem przedwczesnej miażdżycy.
Najcięższy przebieg obserwuje się u pacjentów z wariantami homozygotycznymi lub złożonymi heterozygotycznymi, u których stężenie LDL-C może być skrajnie wysokie już w dzieciństwie.
Do najważniejszych genów związanych z rodzinną hipercholesterolemią (FH) należą:
- LDLR (kodujący receptor LDL) – jego mutacje stanowią najczęstszą przyczynę FH i odpowiadają za zdecydowaną większość przypadków autosomalnej dominującej postaci choroby;
- APOB – mutacje prowadzą do obniżonego powinowactwa LDL (zwłaszcza apoB-100) do receptora LDL, co upośledza wychwyt LDL z krwi;
- PCSK9 – mutacje typu gain-of-function nasilają degradację receptorów LDL w hepatocytach, zmniejszając ich liczbę na powierzchni komórek;
- LDLRAP1 – rzadki gen odpowiedzialny za autosomalnie recesywną postać FH (ARH).
Przyczyny poligenowe hipercholesterolemii
Hipercholesterolemia poligenowa wynika z sumarycznego działania wielu wariantów genetycznych o niewielkim efekcie, które łącznie wpływają na metabolizm LDL i prowadzą do jego przewlekłego podwyższenia. Nasilenie zaburzeń jest zmienne i zależy od liczby oraz kombinacji wariantów genetycznych, co określa się jako skumulowane ryzyko poligenowe.
Fenotyp lipidowy ujawnia się w dużej mierze w interakcji z czynnikami środowiskowymi. Istotne znaczenie mają dieta bogata w tłuszcze nasycone i cholesterol, nadwaga lub otyłość oraz niska aktywność fizyczna, które mogą istotnie nasilać ekspresję genetycznej predyspozycji.
W porównaniu z monogenową postacią rodzinnej hipercholesterolemii zaburzenia lipidowe w hipercholesterolemii poligenowej mają zwykle łagodniejszy i mniej jednorodny charakter, jednak w dłuższej perspektywie mogą istotnie zwiększać ryzyko chorób sercowo-naczyniowych.
Przyczyny hipercholesterolemii wtórnej (nabytej)
Hipercholesterolemia wtórna rozwija się w przebiegu innych chorób, działania leków lub zaburzeń metabolicznych, które wpływają na gospodarkę lipidową. Może występować jako samodzielna przyczyna podwyższonego stężenia LDL-cholesterolu lub ujawniać albo nasilać istniejącą predyspozycję genetyczną do dyslipidemii.
- Czynniki środowiskowe i styl życia:
- dieta bogata w tłuszcze nasycone i izomery trans kwasów tłuszczowych;
- nadmierna podaż kalorii oraz dieta wysoko przetworzona;
- nadwaga i otyłość, szczególnie typu trzewnego;
- mała aktywność fizyczna;
- palenie tytoniu.
Nadmierne spożycie alkoholu nie stanowi typowej przyczyny izolowanego wzrostu LDL-cholesterolu, jednak może znacząco wpływać na zaburzenia lipidowe, przede wszystkim poprzez zwiększenie stężenia triglicerydów.
Czynniki psychologiczne, np. przewlekły stres, działają głównie pośrednio poprzez modyfikację zachowań zdrowotnych i nie stanowią samodzielnej, jednoznacznie udokumentowanej przyczyny hipercholesterolemii.
Do najczęstszych chorób powodujących wtórny wzrost LDL-cholesterolu należą:
- cukrzyca typu 2 oraz insulinooporność (często w ramach zespołu metabolicznego) – prowadzą do dyslipidemii aterogennej, w której obserwuje się podwyższenie stężenia triglicerydów oraz obniżenie HDL, a także obecność małych, gęstych LDL; stężenie LDL bywa prawidłowe lub umiarkowanie podwyższone, przy czym istotniejsza klinicznie jest jego zwiększona aterogenność (zdolność do sprzyjania rozwojowi miażdżycy, czyli odkładania się lipidów w ścianie naczyń i tworzenia blaszek miażdżycowych) niż bezwzględny poziom;
- niedoczynność tarczycy – zmniejsza ekspresję receptorów LDL w wątrobie, co prowadzi do wzrostu stężenia LDL i cholesterolu całkowitego;
- przewlekła choroba nerek oraz zespół nerczycowy – prowadzą do wtórnych zaburzeń lipidowych, które obejmują często istotne zwiększenie stężenia LDL-cholesterolu i triglicerydów; mechanizm ten wiąże się m.in. ze wzmożoną syntezą lipoprotein w wątrobie oraz utratą białek regulujących metabolizm lipidów;
- cholestatyczne choroby wątroby – upośledzają wydalanie cholesterolu z żółcią, co prowadzi do hipercholesterolemii (często z obecnością nieprawidłowych frakcji lipoprotein);
- zespół Cushinga oraz inne zaburzenia endokrynologiczne (np. nadmiar hormonu wzrostu, a w wybranych sytuacjach także androgenów) – mogą prowadzić do insulinooporności, nasilenia lipolizy oraz zwiększonej syntezy VLDL w wątrobie, co skutkuje rozwojem dyslipidemii mieszanej.
Ponadto istotne znaczenie w rozwoju dyslipidemii może mieć zakażenie HIV, a także stosowana terapia antyretrowirusowa, które w odmiennych mechanizmach wpływają na metabolizm lipidów, prowadząc do zaburzeń ich profilu. Zmiany te obserwuje się również w stanach fizjologicznych, takich jak ciąża, w której dochodzi do przejściowego wzrostu stężeń lipidów w ramach adaptacji metabolicznej organizmu (parametry te zazwyczaj ulegają normalizacji po porodzie).
Do leków, które mogą powodować wzrost stężenia LDL-cholesterolu, należą:
- glikokortykosteroidy – powodują wzrost stężenia cholesterolu całkowitego, LDL oraz triglicerydów, zwłaszcza przy długotrwałym stosowaniu; mechanizm obejmuje m.in. nasilenie insulinooporności, zwiększoną lipolizę i wpływ na syntezę VLDL w wątrobie;
- retinoidy (np. izotretynoina) – prowadzą często do istotnego wzrostu triglicerydów oraz (niekiedy) wzrostu LDL i cholesterolu całkowitego; rzadziej obserwuje się zmiany HDL;
- inhibitory mTOR (sirolimus, ewerolimus) – często powodują istotne zaburzenia lipidowe, zwłaszcza wzrost LDL i triglicerydów; wymagają regularnej kontroli lipidogramu;
- leki immunosupresyjne (cyklosporyna, takrolimus) – mogą zwiększać stężenie LDL i cholesterolu całkowitego poprzez zmniejszenie klirensu LDL w wątrobie oraz wpływ na metabolizm lipidów;
- leki przeciwpsychotyczne II generacji, takie jak np. olanzapina czy klozapina – mogą zwiększać stężenie triglicerydów, cholesterolu całkowitego oraz LDL-cholesterolu;
- niektóre β-blokery (zwłaszcza nieselektywne, starsze generacje) – mogą obniżać HDL oraz nieznacznie zwiększać triglicerydy; efekt jest zwykle słaby lub minimalny w przypadku beta-blokerów nowszej generacji (kardioselektywnych i/lub wazodylatacyjnych);
- inhibitory proteazy stosowane w leczeniu HIV – mogą zwiększać stężenie triglicerydów, cholesterolu całkowitego oraz LDL-cholesterolu wskutek zaburzeń metabolizmu lipidów i nasilenia insulinooporności; długotrwała terapia może zwiększać ryzyko sercowo-naczyniowe;
- wybrane preparaty hormonalne (wpływają na profil lipidowy w zależności od dawki, składu, drogi podania) – estrogeny, zwłaszcza doustne, zwykle obniżają LDL-cholesterol i mogą zwiększać stężenie triglicerydów poprzez nasilenie syntezy VLDL w wątrobie; część progestagenów jest z kolei neutralna, ale niektóre z nich (zwłaszcza te o działaniu androgenizującym) mogą pogarszać profil lipidowy.
Kto jest najbardziej narażony na hipercholesterolemię?
Ryzyko rozwoju hipercholesterolemii wzrasta istotnie u osób, u których współwystępuje kilka czynników metabolicznych, środowiskowych lub genetycznych – im większa ich liczba, tym wyższe prawdopodobieństwo podwyższonego stężenia cholesterolu LDL oraz rozwoju powikłań sercowo-naczyniowych w przyszłości. Szczególną grupę stanowią pacjenci z rodzinną hipercholesterolemią.
Objawy hipercholesterolemii
Hipercholesterolemia przez długi czas przebiega bezobjawowo, dlatego wielu pacjentów nie jest świadomych podwyższonego stężenia cholesterolu aż do momentu wykonania badań laboratoryjnych.
Hipercholesterolemia – objawy bezpośrednie (rzadkie)
Widoczne objawy hipercholesterolemii występują stosunkowo rzadko i dotyczą głównie ciężkich postaci zaburzeń lipidowych, zwłaszcza rodzinnej hipercholesterolemii.
Do najbardziej charakterystycznych należą:
- żółtaki ścięgniste (xanthomata tendinea) – złogi cholesterolu odkładające się w obrębie ścięgien, najczęściej ścięgna Achillesa oraz ścięgien prostowników dłoni;
- kępki żółte powiek (xanthelasma palpebrarum) – miękkie, żółtawe zmiany w obrębie powiek; mogą współwystępować z zaburzeniami lipidowymi, ale pojawiają się również u osób z prawidłowym stężeniem cholesterolu;
- rąbek rogówkowy (arcus cornealis) – szaro-biała obwódka na obwodzie rogówki; u młodych osób może sugerować obecność zaburzeń lipidowych, zwłaszcza rodzinnej hipercholesterolemii.
Objawy powikłań miażdżycy związanej z hipercholesterolemią
W większości przypadków pierwsze objawy kliniczne związane z hipercholesterolemią wynikają z rozwoju miażdżycy i jej powikłań sercowo-naczyniowych. Długotrwale podwyższone stężenie cholesterolu LDL sprzyja odkładaniu lipidów w ścianach tętnic, co prowadzi do ich zwężenia oraz upośledzenia przepływu krwi.
Najczęstsze objawy obejmują:
- ból w klatce piersiowej (dławicę piersiową) związany z niedokrwieniem mięśnia sercowego;
- duszność oraz obniżoną tolerancję wysiłku występujące w przebiegu choroby wieńcowej lub niewydolności serca;
- ból kończyn dolnych podczas chodzenia (chromanie przestankowe) związany z miażdżycą tętnic obwodowych;
- ostre incydenty sercowo-naczyniowe (zawał serca lub udar mózgu), które u części pacjentów mogą być pierwszą manifestacją hipercholesterolemii.
Bibliografia
- Arent-Piotrowska K., Hipercholesterolemia – zmora dzisiejszych czasów. Co zrobić, by zapobiec jej konsekwencjom w świetle aktualnych zaleceń kardiologicznych. Probl Hig Epidemiol. 2018; 99(2):108–113.
- Myśliwiec M., Bandura M., Wołoszyn-Durkiewicz A., Hennig M., Walczak M., Peregud-Pogorzelski J., Sykut-Cegielska J., Miszczak-Knecht M., Chlebus K., Wasąg B., 2024 Polish recommendations for the management of familial hypercholesterolemia in children and adolescents, 2025.
- Niemczuk P., Kałużny S., Musialik K., Hipercholesterolemia rodzinna – podłoże genetyczne. Forum Zaburzeń Metabolicznych. 2022;13(4):115–120.
- Rynkiewicz A. i wsp: Postępowanie w heterozygotycznej hipercholesterolemii rodzinnej, Stanowisko Forum Ekspertów Lipidowych, 2013.
- Szymański F. M., Hipercholesterolemia jako najbardziej rozpowszechniony czynnik ryzyka chorób układu sercowo-naczyniowego w Polsce. O czym warto pamiętać w codziennej praktyce? Choroby Serca i Naczyń. 2014;11(4):204–211.
Artykuły z kategorii hipercholesterolemia
- Czym jest dyslipidemia i kiedy zagraża zdrowiu?
- Dieta na obniżenie trójglicerydów – co jeść, a czego unikać?
- Statyny – czym są, jak działają, skuteczność i skutki uboczne
- Triglicerydy (trójglicerydy) – czym są, kiedy badać, jak interpretować wyniki?
- Cholesterol: całkowity, HDL, LDL, nie-HDL – co oznacza, normy


